- 标签
- 热门文章
- 推荐文章
Gaussian 关于体系自旋多重度的确定
设置电荷和多重度是进行量子化学计算很基础的一个部分。但是很多入门的小伙伴也不是很清楚。这里就讲一下大概的设置方法。
电荷∶这个是指当前被算的分子所带的电量。这个是根据需要来自己设定的。而不是去分析分子内的原子的化合价什么。这个完全不同的概念。
比如要算H2如果不告诉高斯带电量,高斯根本不知道要算什么:
设置电荷+1高斯就会计算H2+阳离子;
设置电荷0高斯就会计算H2分子;
设置电荷-1高斯就会计算H2-阴离子。
自旋多重度∶定义是2S+1,S是总自旋角动量。
一个Alpha电子的自旋角动量是+1/2,一般表示成↑;
一个Beta电子的自旋角动量是-1/2,一般表示成↓。
判断一个体系自旋多重度的最基础方法和最本质的方法就是数出体系中Alpha电子个数N(α)和Beta电子的个数N(β)。
自旋多重度=2*{(+1/2)*N(α)+(-1/2)*N(β)}+1=N(α)-N(β)+1
这样做虽然确保没有错,但是很麻烦实际判断中也会有一些简化的判断方法。
1.判断自旋多重度之前必须要先判断体系的总电子数目。
体系总电子数目=体系中原子中的电子数总和-预设好的电荷量。比如H2分子总电子数目=2,H2+总电子数目=1,H2-总电子数=3。
因为一般的体系中绝大部分分子轨道中电子都是Alpha和Beta成对存在的,他们的自旋相反,自旋所以会相互"抵消",所以如果一个体系的总电子数是偶数,那么大概率所有分子轨道中电子都是成对存在的,也就是闭壳的分子,N(α)=N(β),自旋多重度=1。如果一个体系中总电数是奇数,被2除余1,也就是体系中大概率会有一个单独的不成对电子会提供+1/2的自旋。那根据公式计算自旋多重度=2。以上适用于绝大部分有机小分子,简单自由基,生物分子的体系。这也是GaussView默认的判断自旋多重度方法。
2.对于单个原子或者一些简单的小分子如果已经知道了电子在分子轨道中的排布,就很好去判断自选多重度。比如下图两种状态的O2分子。
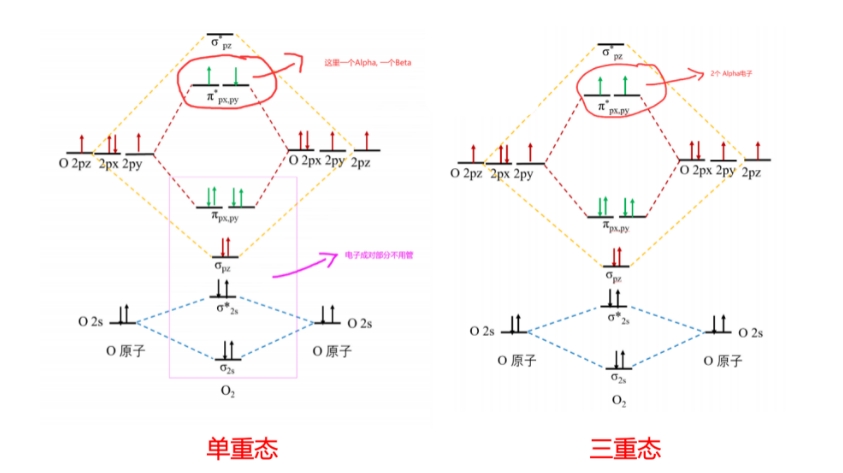
这里特别要注意计算单个原子时候的多重度设置。一定要根据原子的核外电子排序自己修改自旋多重度,因为GaussView默认会使用上一条中提到的方法(偶数电子,单重态;奇数电子,二重态)给出默认的多重度。这个一般对于原子的基态计算都是不正确的。比如C原子是6个电子,是偶数。高斯会默认单重态。但是C原子的基态电子排布是ls²2s2p²两个p电子是平行存在的,自旋方向相同。所以C原子的基态应该是3重态。
3.对于含有过渡金属,重金属的分子是最麻烦的,因为d轨道,f轨道的加入,用总电子数是奇数偶数去判断自旋多重度基本都是没用的。而且分子轨道复杂,也没法一下判断出来电子的排布。这时候就得考虑所有可能性。
比如计算出整个体系的总电子数是偶数,那么体系中不成对电子数可能是0,2,4…
假设它们都是自旋方向都是相同的,那么对应的可能的多重度就是1,3,5……
对应的如果总电子数是奇数,体系中不成对电子数目可能是1,3,5…
对应的可能的多重度就是2,4,6.…….
下一条:Gaussian软件搜索过渡态




 沪公网安备31011302006932号
沪公网安备31011302006932号